


Получено 5 августа 2016 г.; получено с поправками 10 февраля 2017 г.; принято в печать 6 марта 2017 г.; доступно онлайн с 24 марта 2017 г.
Читать статью в формате PDF: Глобальная эпидемиология подтипов вируса гепатита C и мутаций резистентности по результатам секвенирования
Актуальность и цели. Генотип, подтип вируса гепатита С (HCV) и наличие мутаций резистентности (МР) являются основными факторами, определяющими выбор схем противовирусных препаратов прямого действия (ПППД). Однако существующие в настоящее время методы генотипирования HCV имеют недостатки в отношении дифференцировки между подтипами HCV, а распространенность МР практически неизвестна. Целью настоящего исследования было изучить эпидемиологию HCV с помощью 12 645 образцов, взятых у пациентов в 28 странах в 5 географических регионах.
Методы. Мы сравнивали генотип и подтипы HCV с помощью метода INNO-LiPA 2.0 и метода секвенирования ампликона у 8945 участников клинических исследований ПППД II и III фаз. Изучена глобальная молекулярная эпидемиология HCV у 12 615 пациентов. Распространенность МР изучалась с помощью популяционного или глубокого секвенирования, также был выполнен филогенетический анализ для исследования разнообразия подтипов.
Результаты. Несмотря на высокую согласованность между методами INNO-LiPA и секвенирования при определении генотипа, метода INNO-LiPA было недостаточно для определения генотипов 2, 3, 4 и 6. Секвенирование позволило уточнить наличие генотипов 2, 3, 4 и 6 у 42, 10, 81 и 78 % пациентов соответственно. Несовпадение результатов определения генотипа с помощью разных методов (генотип 2 — генотип 1) наблюдалось у 28 (3 %) из 950 пациентов с генотипом 2, что согласуется с наличием рекомбинаций между генотипами.
Анализ результатов секвенирования продемонстрировал различия в распространенности подтипов в разных регионах, особенно генотипов 2, 4 и 6. Распространенность МР варьировала в зависимости от подтипа. При этом клинически значимая мутация гена NS3 Q80K была обнаружена у пациентов с генотипами 1а, 5а и 6а, а мутация гена NS5A Y93H была выявлена у больных с генотипами 1b, 3а, 4b, 4r и 7.
Выводы. Результаты данных анализов дают представление о точности диагностики подтипа и распределении МР, что имеет ключевое значение для внедрения глобальных стратегий лечения гепатита C.
Резюме. HCV обладает высокой изменчивостью. На сегодня описано 7 его генотипов и 67 подтипов. Целью настоящего исследования было: 1) сравнить два разных метода для определения генотипов; 2) изучить распространенность подтипов HCV каждого генотипа во всем мире; 3) установить распространенность МР внутри разных подтипов.
Мы обнаружили, что оба метода характеризуются высокой согласованностью при разделении генотипов, однако определение конкретного подтипа было не всегда точным. Анализ результатов секвенирования показал наличие разницы в распространенности подтипов некоторых генотипов по регионам, особенно подтипов генотипов 2, 4 и 6. Распространенность МР также колебалась в зависимости от подтипа. На основании этих различий можно судить об эффективности разных препаратов для лечения гепатита C.
Сокращения: GT — генотип; HCV — вирус гепатита С; LDV — ледипасвир; PegIFN — пегилированный интерферон; RBV — рибавирин; SOF — софосбувир; UTR — нетранслируемая область; МР — мутации резистентности; ПППД — противовирусные препараты прямого действия; УВО — устойчивый вирусологический ответ.
Введение
Вирус гепатита С (HCV) является основным возбудителем инфекционного гепатита у человека. Установлено, что в мире HCV-инфекцией страдает более 170 млн человек, для которых характерен повышенный риск развития прогрессирующего поражения печени и гепатоцеллюлярного рака [1, 2]. HCV классифицируется на 7 генотипов и 67 подтипов и характеризуется дивергенцией около 30 и 15 % на уровне подтипов соответственно [3].
Разнообразие HCV отчасти обусловлено мутациями в гене РНК-зависимой РНК-полимеразы (RdRp), у которого отсутствует способность к исправлению ошибок считывания, а также рекомбинацией, дрейфом генов и иммунным давлением на вирус [4]. В лечении HCV-инфекции произошла революция после внедрения высокоэффективных противовирусных препаратов прямого действия (ПППД), которые направлены на такие генные продукты HCV, как NS5B, фосфопротеин NS5A и протеаза NS3 [5].
Так же как и при лечении пегилированным интерфероном (PegIFN) и рибавирином (RBV), выбор схемы ПППД, продолжительность лечения и устойчивый вирусологический ответ (УВО) зависят от генотипа и подтипа HCV [6–9]. В предыдущих исследованиях было показано, что распределение генотипов HCV варьирует во всем мире [10, 11], хотя относительно распределения подтипов известно меньше.
Обнаружено, что подтипы HCV группируются на филогенетическом дереве в зависимости от региона происхождения [12–14]. Потенциально увеличение разнообразия HCV в локальном эпидемическом процессе может повлиять на частоту достижения УВО при возникновении мутаций резистентности (МР).
Распространенность в мировом масштабе рекомбинантных форм HCV, обнаруженных в Восточной Европе и России [15–17], пока неизвестна. Выявление рекомбинантных последовательностей позволит оценить глобальную распространенность известных рекомбинантных вирусов, таких как RF2k/1b, и, возможно, обнаружить новые рекомбинантные формы в популяции.
Получение данных относительно распространенности генотипов и подтипов вируса во всем мире имеет большое значение, т. к. позволит оптимизировать выбор схем терапии и продолжительности лечения.
Систематическая оценка МР к ПППД, направленным на вирусные белки NS3, NS5A и NS5B, для разных генотипов HCV не проводилась [18, 19]. МР варьируют на уровне подтипов [20] и при наличии до начала лечения могут повлиять на его результаты. Таким образом, в условиях отсутствия секвенирования для каждого пациента целесообразно оценивать МР с учетом подтипа HCV для выбора оптимальной схемы ПППД.
Целью настоящего исследования было изучить эпидемиологию HCV с помощью анализа 125 615 образцов, полученных у пациентов в 28 странах в 5 географических регионах. Поскольку точность существующих в настоящее время методов генотипирования может различаться, сначала мы сравнили классификацию генотипов с помощью метода INNO-LiPA, который является современным стандартом [21, 22] и используется для секвенирования ампликонов NS3/4A, NS5A и NS5B, в выборке из 8945 образцов. Затем мы изучили распространенность подтипов HCV и специфических для конкретных подтипов МР в разных географических регионах.
Материалы и методы
Клинические образцы
Для участия в 67 исследованиях, проведенных компаниями Gilead Sciences (США) и Pharmasset (США), информированное согласие было подписано 12 615 пациентами. Исследования и число участников приведены в дополнительной табл. S1. Протоколы исследований соответствовали этическим принципам Хельсинкской декларации и были одобрены этическими комитетами соответствующих медицинских центров.
Все образцы, включенные в настоящий анализ, были взяты в начале исследований у пациентов, не получавших ранее терапии, и у пациентов, уже получавших ранее лечение. Более 95 % включенных пациентов ранее не получали ПППД.
Генотипирование с помощью методов INNO-LiPA и секвенирования ампликона
Анализ с помощью метода Siemens VERSANT HCV Genotype INNO-LiPA 2.0 (Innogenetics, Бельгия) выполнялся в лабораториях Covance (США, Сингапур, Швейцария), QLabs (Великобритания, США, Сингапур) и Cenetron (США). Генотип, определенный с помощью метода INNO-LiPA, использовался для выбора генотип-специфических праймеров, расположенных за пределами генов-мишеней, амплифицирующих регионы NS3/4A, NS5A и NS5B в образцах пациентов.
В зависимости от клинического исследования у 1 пациента могло быть несколько амплифицированных генов-мишеней. Стандартная полимеразная цепная реакция с обратной транскрипцией с использованием плазмы пациентов с уровнем РНК HCV > 1000 МЕ/мл выполнялась в диагностической лаборатории DDL (Нидерланды).
Если амплификация с помощью генотип-специфических праймеров оказывалась безуспешной, частичную последовательность NS5B амплифицировали с помощью генотип-независимых праймеров, что позволяло выполнить классификацию генотипа/подтипа.
При глубоком секвенировании ампликоны, кодирующие белки NS3/4A, NS5A и NS5B, обрабатывали путем глубокого секвенирования 150 парных концов с помощью системы Illumina MiSeq v2 в лаборатории DDL или WuXi AppTec (Китай). Файлы FASTQ разделяли на основании 100 % совпадений штрих-кодов. Непрерывные последовательности формировали из файлов FASTQ парных концов с помощью программы VICUNA (Broad Institute, США) и соединяли для создания последовательности denovo.
Все прочтения парных концов соединяли с помощью программы PEAR (Exelixis Lab, Германия), разбивали в области 3′-конца при MAPQ < 15, и фильтровали для удаления прочтений с менее 50 пар оснований. Профильтрованные прочтения выравнивали до соединения последовательности de novo с помощью программы MOSAIK v1.1.0017 (Marth Lab, США) для создания окончательной соединенной последовательности.
Выровненные последовательности транслировали внутри рамки, а полученную в результате таблицу вариантов окончательной последовательности использовали для создания консенсусной последовательности. О смесях сообщалось при наличии не менее 15 % вирусной популяции. Последовательности популяции охватывали варианты с распространенностью 15 %.
С целью определить генотип или подтип HCV консенсусные нуклеотидные и аминокислотные последовательности NS3/4A, NS5A и NS5B сравнивали путем выравнивания с помощью программы BLAST на NCBI [23] до набора эталонных последовательностей. В целом для сравнения было использовано 170 NS3, 167 NS5A и 169 NS5B эталонных последовательностей, представляющих 7 генотипов и 67 подтипов.
В этом эталонном наборе каждый подтип был представлен 1–7 последовательностями. Генотип или подтип считались установленными, если их нуклеотидные или аминокислотные последовательности были на 85 % гомологичными эталонной последовательности. Порог 85 % был выбран, чтобы определить подтип и избежать неправильной оценки.
В каждом образце устанавливали подтип, наиболее гомологичный последовательности на нуклеотидном или аминокислотном уровне. Если секвенировали множество мишеней, в результате анализа последовательности пациента определялось множество подтипов.
В табл. 1 образец обозначался как новый/смешанный при отсутствии гомологичности минимум 85 % с наиболее близкой эталонной последовательностью, если генотип и подтип, определенные на основании гомологичности аминокислотной и нуклеотидной последовательностям, не были идентичными или если подтипы не были идентичными между ампликонами.
В табл. 2 и на рис. 1 только ампликон NS5B использовался для определения подтипа. Образец обозначался как новый/смешанный при отсутствии гомологичности минимум 85 % с наиболее близкой эталонной последовательностью или если основные аминокислотные и нуклеотидные подтипы не были идентичными.
Считалось, что в образце содержится рекомбинантный вирус GT2-GT1, если анализ INNO-LiPA показывал наличие вируса GT2, тогда как секвенирование NS3/4A, NS5A и/или NS5B показывало наличие вируса GT1, а при полногеномном секвенировании обнаруживался точечный разрыв при рекомбинации вируса [24].
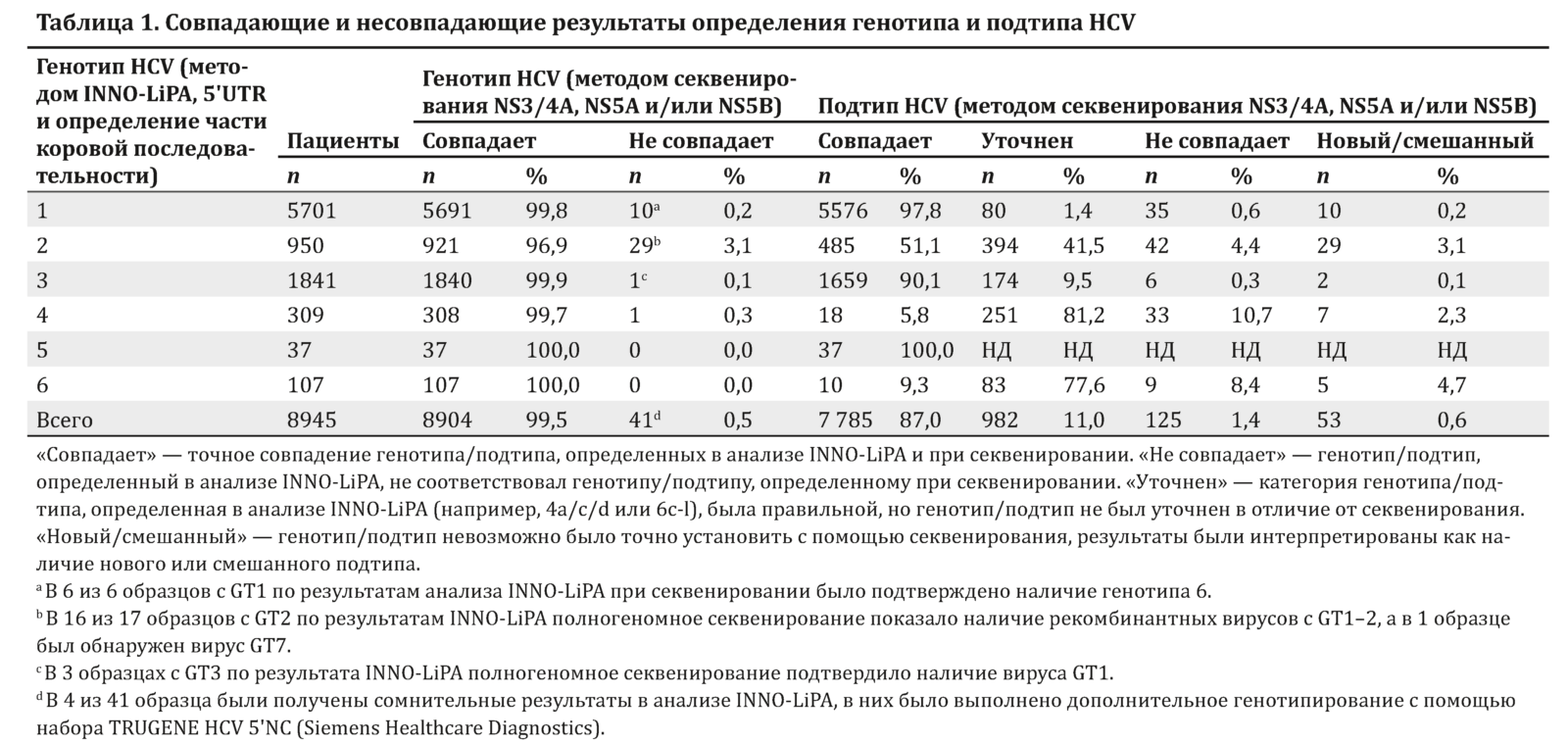
Совпадение результатов INNO-LiPA и секвенирования
Результаты сравнения генотипа и подтипа, определенных методами INNO-LiPA и секвенирования у 8945 пациентов на момент начала исследования, были внесены в табл. 1, где они были разделены на категории, как совпадающие и не совпадающие на уровне генотипа. На уровне подтипов результаты были совпадающими, когда результаты, полученные в анализе INNO-LiPA, соответствовали результатам секвенирования, например GT2a в анализе INNO-LiPA и GT2a при секвенировании.
Результат считался установленным, если правильный генотип или подтип определялся в анализе INNO-LiPA, но не был точным, например GT2 или GT2a/2c в анализе INNO-LiPA и GT2a при секвенировании. Результат был несовпадающим, если в анализе INNO-LiPA был определен неправильный генотип или подтип по сравнению с секвенированием. Выявленный генотип/подтип считался новым/смешанным, если при секвенировании невозможно было установить точный подтип.
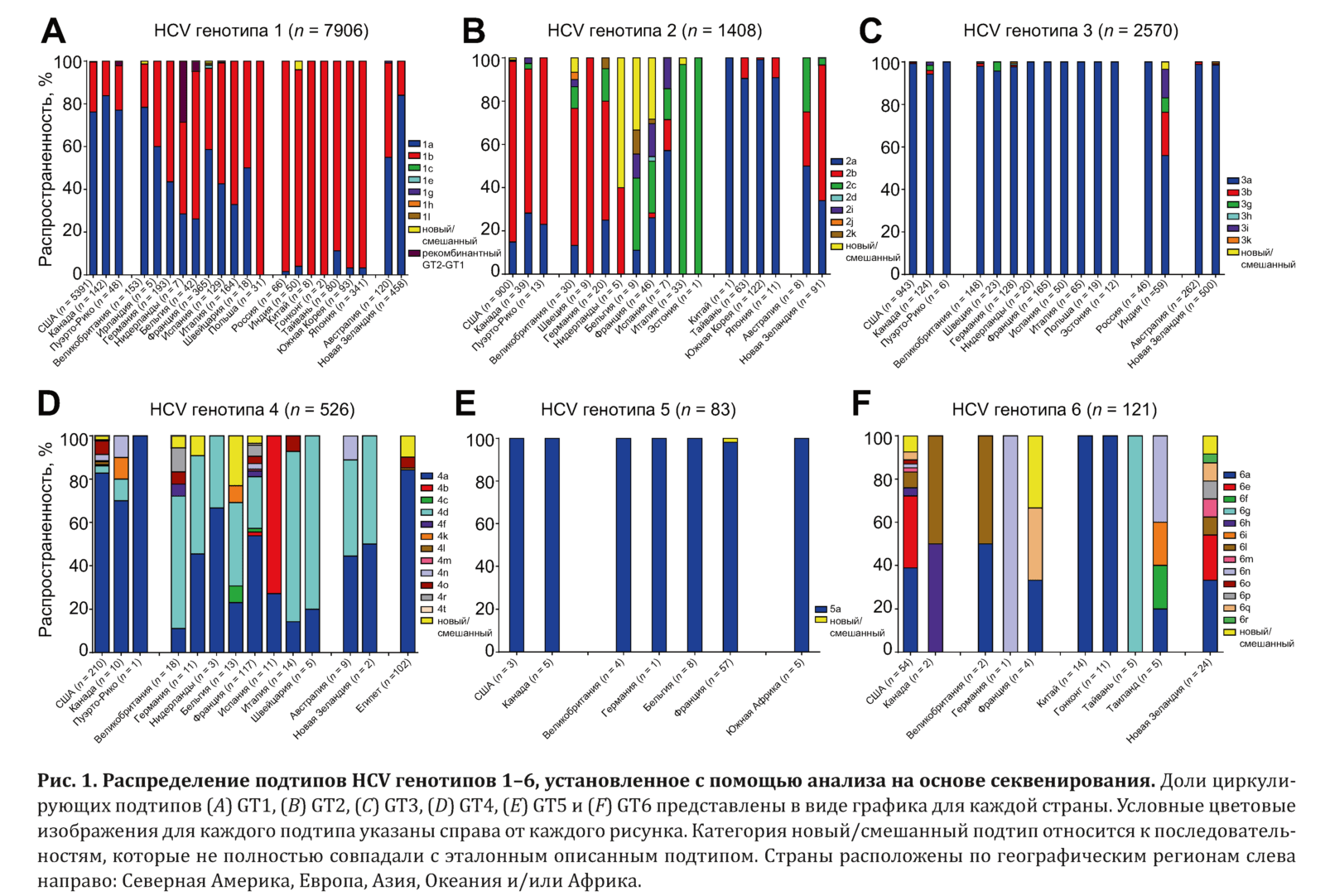
Глобальная распространенность подтипов HCV
Распространенность подтипов HCV оценивалась на основании их определения с помощью секвенирования NS5B в начале исследования у 12 615 пациентов и изучения стран их происхождения. В табл. 2 и на рис. 1 вирус рассматривался как новый/смешанный, если нуклеотидная или аминокислотная последовательность NS5B, определенная с помощью программы BLAST, не соответствовала эталонной последовательности.
Распространенность подтипа рассчитывалась следующим образом: число подтипов в стране/число генотипов в стране. Регионы и страны, в которых изучалась распространенность: Северная Америка (США, Канада, Пуэрто-Рико), Европа (Великобритания, Ирландия, Швеция, Германия, Нидерланды, Бельгия, Франция, Швейцария, Польша, Эстония), Азия (Россия, Индия, Китай, Гонконг, Тайвань, Корея, Япония, Таиланд), Океания (Австралия, Новая Зеландия) и Африка (Египет, Южная Африка).
Последовательности NS5B, представляющие генотипы 1c, 1e, 1g, 1h, 1l, 2i, 2j, 2k, 3g, 3i, 4c, 4f, 4k, 4l, 4n, 4o, 4r, 4t, 6e, 6f, 6h, 6i, 6l, 6m, 6n, 6p и 6q, были сохранены в базе данных GenBank (KY608611-KY608718).
Распространенность мутаций резистентности в зависимости от подтипа HCV
Распространенность МР в подтипах HCV определялась у пациентов для генов NS3 (n = 2664), NS5A (n = 7893) и NS5B (n = 11 074) в начале исследования. Пациенты, уже получавшие ранее ПППД, исключались из анализа. Для каждого подтипа выполняли выравнивание аминокислотных последовательностей ампликона пациента и анализировали расположение МР с определением частоты наличия аминокислот в указанном месте.
МР выявлялись при распространенности не менее 15 % у пациента. Из зарегистрированных МР мутации с распространенностью не менее 5 % в популяциях с определенным подтипом указаны в табл. 3–5. Наиболее и наименее распространенные варианты для каждого подтипа регистрировались по отношению к GT1a H77 эталонной аминокислоты в указанном положении.

Филогенетический анализ
У пациентов с инфекцией, вызванной генотипами GT3a и GT1b, были выполнены индивидуальные филогенетические анализы для последовательности NS5A. Последовательности выравнивали с помощью программы ClustalW [25]. Филогенетическое дерево для NS5A GT3a включало 468 последовательностей (Австралия: n = 55; Канада: n = 28; Германия: n = 43; Франция: n = 50; Англия: n = 34; Индия: n = 33; Италия: n = 26; Новая Зеландия: n = 60; Россия: n = 69; США: n = 70). Филогенетическое дерево для NS5A GT1b включало 738 последовательностей (Австралия: n = 36; Канада: n = 7; Чехия: n = 10; Англия: n = 11; Франция: n = 34; Германия: n = 96; Индия: n = 46; Италия: n = 67; Япония: n = 70; Корея: n = 83; Новая Зеландия: n = 35; Нидерланды: n = 1; Польша: n = 29; Испания: n = 22; Тайвань: n = 68; США: n = 123).
Максимально вероятные филогенетические деревья создавались с помощью параметров, используемых по умолчанию в программе GARLi (версия 2.0), что позволяет оптимизировать подстановочную модель [26]. Статистическая значимость ветвей оценивалась с помощью теста отношения приближенного правдоподобия [27] с использованием программы PhyML 3.0 [28], а консенсусное дерево создавалось с помощью программы PAUP (версия 4.0b10). Ветви со значением p < 0,001 считались подтвержденными.
Филогенетические деревья наглядно представляли с помощью программы FigTree (версия 1.3.1). Кластер определяется как филогенетически связанные последовательности, которые отделены от всех остальных последовательностей значительной ветвью.






Результаты
Сравнение генотипа и подтипа с помощью анализа INNO-LiPA и секвенирования ампликона
Анализ INNO-LiPA и секвенирование ампликона были выполнены у 8945 HCV-инфицированных пациентов с целью определить генотип (GT) и подтип HCV. Согласно результатам анализа INNO-LiPA, у 5701 пациента был GT1, у 950 — GT2, у 1841 — GT3, у 309 — GT4, у 37 — GT5 и у 107 — GT6.
Между двумя методами отмечался высокий уровень согласованности, как показано в табл. 1. При определении GT1 (99,8 %), GT3 (99,9 %), GT4 (99,7 %), GT5 (100 %) и GT6 (100 %) согласованность между двумя методами составляла более 99 %, тогда как при установлении GT2 она была равна 96,9 %. В целом генотипы, определенные в анализе INNO-LiPA и при секвенировании ампликона, были одинаковыми у 8904 (99,5 %) из 8945 пациентов.
Частота согласованности при определении GT2 была примерно на 3 % ниже, чем при установлении всех остальных генотипов. У 29 пациентов в образцах был предположительно неверно установлен генотип GT2 с помощью анализа INNO-LiPA. В 28 из этих 29 несовпадающих образцов во время секвенирования ампликона был выявлен GT1.
Из этих 29 несовпадающих образцов 17 были изучены с помощью полногеномного секвенирования. В 16 из этих 17 случаев HCV представлял собой рекомбинацию GT2 (5′UTR-NS2) и GT1 (NS3-3′UTR), с точкой разрыва на границе NS2-NS3, как было описано ранее [29]. 6 из этих 16 рекомбинантных вирусов были идентифицированы как циркулирующий штамм RF2k/1b и были обнаружены у пациентов из Бельгии (2/6), Нидерландов (2/6), Испании (1/6) и США (1/6). Другие 6 из 16 рекомбинантных вирусов GT2-GT1 представляли собой рекомбинантный штамм GT2b/1a и были обнаружены у пациентов из США.
Остальные 4 рекомбинантных вируса GT2-GT1 относились к рекомбинантному штамму GT2b/1b и были выделены у пациентов из США (3/4) и Пуэрто-Рико (1/4). 12 из этих 16 рекомбинантных вирусов GT2-GT1 были описаны ранее [29]. Интересно, что в оставшемся образце GT2 с несовпадающими результатами (1/17) был обнаружен вирус с GT7 [30].
Изучались также образцы с несовпадающими результатами, в которых с помощью анализа INNO-LiPA были выявлены генотипы GT1, 3 и 4. Из 10 образцов, не совпавших по GT1, при секвенировании ампликона был обнаружен GT6. При полногеномном секвенировании, выполненном в 6 из этих 8 несовпадающих образцов с GT1, было подтверждено наличие вируса с GT6.
Было только 2 несовпадающих образца, в которых по результатам INNO-LiPA было установлено наличие либо GT3 (n = 1), либо GT4 (n = 1). В обоих случаях секвенирование ампликона показало наличие GT1. Подобно секвенированию ампликона, полногеномное секвенирование, выполненное в несовпадающем образце с GT3 по результатам INNO-LiPA, установило наличие GT1.
На уровне подтипов отмечались более выраженные колебания частоты совпадения результатов между анализом INNO-LiPA и секвенированием ампликона, чем на уровне генотипов. Определение подтипов GT5 и GT1 оказалось наиболее эффективным: 100 и 97,8 % результатов были подтверждены при генотипировании ампликона. Для остальных генотипов отмечался более низкий уровень совпадения подтипов.
Частота совпадения результатов при определении подтипа вируса GT2, 3, 4 и 6 с помощью анализа INNO-LiPA и секвенирования ампликона составляла 51,1, 90,1, 5,8 и 9,3 %, и требовалось уточнение для 41,5, 9,5, 81,2 и 77,6 % образцов соответственно. Для этих генотипов одной из причин уточнения результатов INNO-LiPA является разделение подтипов на группы, как в группах подтипов 2a/2c, 4a/4c/4d и 6c-l.
Частота несовпадения подтипов в тех случаях, когда с помощью анализа INNO-LiPA подтип не был определен, была низкой для GT1 (0,6 %), 3 (0,3 %) и 5 (0 %). Напротив, при исследовании вирусов GT2, 4 и 6 уровень несовпадения был выше и составлял 4,4, 10,7 и 8,4 % соответственно. Более того, среди вирусов с GT2, 4 и 6 отмечалась более высокая частота новых и/или смешанных подтипов (3,1, 2,3 и 4,7 % соответственно).
Географическое распределение подтипов HCV
Распределение подтипов HCV для каждого генотипа анализировали с использованием набора данных, полученных у 12 615 пациентов с 7 генотипами HCV в 28 странах 5 географических регионов, а именно: Северной Америки (n = 7891), Европе (n = 2166), Азии (n = 977), Океании (n = 1474) и Африке (n = 107). Для определения подтипов у всех пациентов использовалось секвенирование NS5B. Настоящий анализ охватывал 46 ранее описанных подтипов HCV GT1–6, в т. ч. 7 GT1, 7 GT2, 6 GT3, 12 GT4, 1 GT5 и 13 GT6.
В подтипы GT1–6 была включена категория новых/смешанных подтипов в связи с выявлением большого числа последовательностей, не соответствовавших ранее описанным штаммам. Кроме того, были изучены рекомбинантные последовательности GT2-GT1 и последовательность GT7. Таким образом, 54 подтипа HCV и их распространенность в мире в зависимости от региона представлены в табл. 2 и на рис. 1.
В нашей выборке образцов с вирусами GT1 (n = 7906) наиболее распространенными подтипами во всех странах были 1а и 1b. В Северной Америке и Океании распространенность подтипа 1а составляла 76,4–78,0 %, а подтипа 1b — 21,8–23,2 %, тогда как в Европе эти показатели равнялись 49,9 и 48,4 %, а в Азии — 4,1 и 95,6 % соответственно (см. табл. 2).
Интересно, что в Польше, которая была включена в анализ в составе Европы, распространенность подтипа 1b соответствовала таковой в Азии и составляла 100 %. Новые/смешанные последовательности GT1 были обнаружены в Великобритании (1,3 %), Франции (0,8 %), Индии (4 %) и, с меньшей частотой, в США (0,1 %).
Рекомбинантные штаммы HCV GT2 (RCF2k-1b, GT2b-1a и GT2b-1b), включенные в категорию GT1, составляли 0,2 % в Северной Америке и 0,5 % в Европе.
В отличие от GT1 циркулирующие подтипы GT2 (n = 1408) широко варьировали. В Северной Америке обнаруживались преимущественно подтипы 2b (82,7 %) и 2a (15,6 %), тогда как в Азии был выявлен главным образом подтип 2a (95,9 %), после которого следовал подтип 2b (4,1 %). В Океании первенство разделили подтипы 2b (59,6 %), 2a (35,4 %) и 2c (5,1 %).
Интересно, что в Европе согласованности в частоте циркулирующих подтипов GT2 не было. В Германии и Великобритании результаты были аналогичны полученным в Океании, с преимущественным выявлением подтипов 2b (55–63,3 %), 2a (13,3–25,0 %) и 2c (10–15 %), за исключением того, что в этой популяции также обнаруживались подтипы 2i, 2j и 2k. В Италии зарегистрирован уникальный профиль подтипов GT2 со 100%-м выявлением подтипа 2с, который в остальных регионах обнаруживался в менее 34 % случаев.
Интереснее всего то, что в Нидерландах (60 %), Бельгии (33,3 %) и Франции (28,3 %) высокая доля циркулирующих подтипов GT2 была представлена новыми/смешанными подтипами. Кроме того, в этих странах были обнаружены циркулирующие подтипы 2a, 2b, 2c, 2i и 2k, что отличает эти страны от других регионов с преимущественным выявлением подтипов 2a, 2b и/или 2c.
Для GT3 (n = 2570) подтип 3a является наиболее распространенным в Северной Америке (98,7 %), Европе (98,9 %) и Океании (98,7 %). Тем не менее отличительной чертой для Азии является снижение распространенности этого подтипа, что обусловлено циркуляцией других подтипов в Индии.
Так, в 55,9 % образцов, взятых в Индии, был обнаружен HCV подтипа 3а, однако в этой популяции также были выявлены и подтипы 3b (20,3 %), 3g (6,8 %), 3i (13,6 %) и не описанный ранее новый/смешанный подтип (3,4 %). Напротив, в России, включенной в анализ в составе Азии, подтип 3а характеризовался 100%-й распространенностью.
Подобно GT2, распределение подтипов GT4 (n = 526) широко варьировало, особенно в Европе. В нашей выборке в Северной Америке преобладал подтип 4а (82,4 %). В Океании циркулировали подтипы 4a (45,5 %), 4d (45,5 %) и 4n (9,1 %). В Африке, которая в настоящем исследовании была представлена образцами, полученными в Египте, выявлялся преимущественно подтип 4a (84,3 %), за которым следовали новые/смешанные подтипы (9,8 %), подтипы 4o (4,9 %) и 4l (1 %).
Примечательно, что новые/смешанные подтипы GT4 были выявлены в Египте по мере распространения HCV в популяции в 1960–1980 гг. во время проведения кампании против шистосомоза [31], что предположительно вызвало распространение инфекции. В Европе распространенность подтипов GT4 изменялась в зависимости от страны, однако при этом преимущественно обнаруживались подтипы 4а и 4d, как в Океании. Однако Испания была единственной страной с циркулирующим подтипом 4d (72,7 %).
В Бельгии (23,1 %), Германии (9,1 %), Великобритании (5,6 %) и Франции (3,4 %) обнаружены высокие уровни циркулирующих новых/смешанных подтипов. Интересно, что новые/смешанные подтипы GT2 также были обнаружены в Бельгии, Великобритании и Франции.
При исследовании GT5 (n = 83) подтип 5а был наиболее распространенным во всех странах. Единственным исключением был 1 пациент во Франции, у которого был выявлен новый/смешанный подтип GT5. Даже с учетом ограниченного количества пациентов с GT6 (n = 121) обнаружено широкое разнообразие подтипов GT6 во всех регионах. Единственным описанным ранее подтипом, который был выявлен в Северной Америке (37,5 %), Европе (33,3 %), Азии (74,3 %) и Океании (33,3 %), был подтип 6а, однако частота его обнаружения широко колебалась. Другим общим признаком между регионами было выявление новых/смешанных подтипов, которые были зарегистрированы в США (7,4 %), Франции (33,3 %) и Новой Зеландии (8,3 %).
Распространение мутаций резистентности среди подтипов HCV
В нашем анализе данных относительно последовательностей пороговое значение 15 % было выбрано с целью определить присутствие полиморфизмов в консенсусной последовательности HCV пациента. Для оценки распространенности МР в различных подтипах мы регистрировали присутствие МР в подтипе, если эта мутация выявлялась в консенсусной последовательности минимум 5 % образцов.
На наличие МР анализировались последовательности NS3 (n = 2664), NS5A (n = 7893) и NS5B (n = 11 074). Исходная частота МР для разных подтипов HCV обобщена для последовательности NS3 в табл. 3. Обнаружена высокая распространенность МР Q80K в последовательности NS3 GT1а по сравнению с GT1b, в связи с чем рекомендуется проведение тестирования на МР Q80K перед началом лечения ингибитором NS3/4A симепревиром из-за снижения частоты УВО при наличии этой мутации [32].
В нашем анализе подтверждена высокая исходная распространенность мутации Q80K в вирусе GT1a (36 %) в отличие от GT1b (1,7 %). Мутация Q80K была также обнаружена в вирусе GT5а (100 %) и GT6a (100 %). В целом в 100 % подтипов GT2 HCV была выявлена мутация Q80G, однако эта замена не была связана с резистентностью. Мутация V36L была обнаружена в большинстве подтипов GT2, 3, 4 и 5 и в 1 последовательности GT7 в нашем наборе данных.
Хотя мутации в подтипах GT1c (n = 1; R155D, A156T, D168E), 1e (n = 2; V36L, T54S, S122N) и 7 (n = 1; V36L, V55A, D168Q) не были представлены широко, в начале исследования было обнаружено минимум 3 МР в последовательности NS3. Другой значимой МР в последовательности NS3 была мутация D168E в подтипах GT5a (53 %) и 6a (7 %).
Исходная частота МР в последовательности NS5A в зависимости от подтипа HCV представлена в табл. 4. По сравнению с NS3 отмечалось более выраженное разнообразие положений МР в последовательности NS5A. Наличие мутации Y93H в последовательности NS5A связано со снижением эффективности ПППД NS5A, с наличием или отсутствием мутации L31M/V/I у пациентов с генотипом GT1b [33].
Исходные мутации Y93H были выявлены у пациентов с HCV генотипов GT1b (11 %), 3a (6 %), 4b (50 %), 4r (13 %) и у 1 с GT7. Другими заменами, которые выявлялись с частотой не менее 50 % в этом положении, были замены Y93F (GT1g), Y93S (GT6f, 6m, 6n) и Y93T (GT6a, 6e, 6f, 6g, 6h, 6l, 6o, 6p, 6q, 6r). Родственная МР L31M/V/I, которая повышает резистентность к ингибиторам NS5A, вызванную мутацией Y93H [34], была обнаружена в подтипах GT1b (4 %), 4b (100 %) и 4r (25 %), но не в подтипе GT3a или 7. Мутация L31M/V/I выявлялась часто в вирусах многих подтипов GT2, 3 и 4. Необходимо отметить, что у всех пациентов с GT4r в начале исследования имелись МР в последовательности NS5A (M28M/V + Q30R + L31M или M28M/V + Q30R + Y93H), у 4 из 8 пациентов было 2 МР в последовательности NS5A (M28M/V/I + Q30R), у 1 из 8 с GT4r — 1 МР в последовательности NS5A (Q30R).
Исходные МР к ингибиторам нуклеозидов в последовательности NS5B были редкими и представлены в табл. 5 в зависимости от подтипа HCV. Мутация S282T, которая обеспечивает резистентность к ингибитору NS5B софосбувиру (SOF) [35], в анализируемых подтипах не была обнаружена в количествах, поддающихся определению.
Разнообразие внутри подтипа
Разнообразие HCV представлено большим числом описанных генотипов и подтипов, как это видно на примере GT4 и 6, а также вариациями внутри подтипов. Как GT1b, так и 3а представляют собой подтипы, которые обнаруживаются с высокой частотой в разных регионах, и с учетом результатов определения подтипов частота их выявления, очевидно, не различается между регионами.
Тем не менее локальная эпидемия может привести к появлению регионального разнообразия подтипов, которые выявляются в филогенетическом анализе на основании образования близкородственных кластеров последовательностей.
Необходимо отметить, что большинство последовательностей GT1b NS5A (рис. 2), полученных у 46 пациентов в Индии (красный), образовывало монофилетические кластеры, а также один кластер со смешанными последовательностями из других стран. При построении филогенетического дерева GT1b NS5A было выявлено образование кластеров из других стран. Япония, Корея и Тайвань (желтый, оранжевый и розовый) были частью двух основных кластеров с 60 и 160 последовательностями, в которых только 27 последовательностей происходили из других стран.
Из 35 последовательностей из Новой Зеландии (голубой) 23 создавали отдельный кластер с 15 последовательностями из Австралии (темно-синий). Как и в случае с GT1b, большинство последовательностей GT3a NS5A, полученных у пациентов в Индии, образовывало значительный кластер (рис. 3).
Обнаружено также формирование кластеров последовательностей GT3a, полученных у пациентов в России (серый). В отличие от Индии российские последовательности GT3a имели три основных кластера, которые образовали группу с последовательностями из Европы и Северной Америки.
Напротив, отмечался высокий уровень смешивания между последовательностями GT1b и 3a в странах Северной Америки (фиолетовый), Европы (зеленый) и Океании (синий). Однако, поскольку анализируемые последовательности были получены у небольшого числа пациентов, принимавших участие в клинических исследованиях, требуется большее число образцов, чтобы установить, являются ли кластеры последовательностей репрезентативными в отношении циркулирующих вирусов в указанных странах.


Обсуждение
Всесторонний анализ точности определения генотипа и подтипа вируса был выполнен, чтобы получить представление о недостатках стандартных методов установления подтипов. В настоящее время стандартным считается метод INNO-LiPA — метод гибридизации с линейными зондами, в котором используются зонды к 5′-нетранслируемой области (UTR) и части коровой последовательности для определения типа HCV [12, 13].
Анализы, направленные на короткую часть генома, могут неправильно установить генотип вируса при высокой гомологичности последовательностей и характеризуются ограниченной способностью при выявлении рекомбинантных и/или новых штаммов [15, 16]. Секвенирование ампликона и расшифровка полного генома HCV являются альтернативными методами, обеспечивающими более высокую точность определения генотипа и подтипа [16].
Однако использование этих методов секвенирования во всех клиниках представляется непрактичным. С учетом географического распределения HCV в некоторых регионах возможна неправильная или неполная идентификация вируса с помощью стандартных методов.
Мы обнаружили высокий уровень совпадения результатов определения генотипов с помощью метода INNO-LiPA и секвенирования ампликона при анализе последовательностей NS3/4A, NS5A и/или NS5B, что согласуется с данными предыдущих исследований [29, 36]. В целом доля несоответствия результатов составляла 0,5 %, причем наибольшая доля несовпадений выпала на GT2. Результаты полногеномного секвенирования несовпадающих образцов с HCV GT2 показали, что несоответствие было обусловлено наличием рекомбинантных форм HCV GT2-GT1 (16 из 17 случаев).
В 6 из 16 рекомбинантных вариантов GT2-GT1 было установлено наличие подтипа RF2k/1b. Известно, что подтип RF2k/1b циркулирует в странах Европы, с высокой распространенностью в Грузии, и впервые был описан у больного из Санкт-Петербурга, Россия [15–17]. В полученных вирусах имелись неопределяемые точечные разрывы, возникшие при рекомбинации, в NS2 из оригинального санкт-петербургского штамма RF2k/1b. Проведенный ранее филогенетический анализ [29] показал, что эти пациенты, вероятно, были инфицированы циркулирующим штаммом RF2k/1b, что не связано с единичными рекомбинациями.
Пациенты с вирусом RF2k/1b выявлены в Европе, в т. ч. в Бельгии (n = 2), Нидерландах (n = 2) и Испании (n = 1), а также в США (n = 1). Первые случаи регистрации штаммов RF2k/1b в Бельгии отмечались у пациентов, прибывших из Грузии и Чехии [37]; однако этническое происхождение 2 пациентов в нашем исследовании неизвестно.
Все не-RF2k/1b рекомбинантные штаммы были получены в Северной Америке, включая 6 пациентов с рекомбинантным вирусом GT2b/1a и 4 пациентов с вирусом GT2b/1b. В отличие от циркулирующего вируса RF2k/1b циркулирующие вирусы GT2b/1a или 2b/1b не были распространенными.
У пациентов с вирусами GT2b/1a или 2b/1b отсутствовал однозначный общий точечный разрыв при рекомбинации. Результаты филогенетического анализа [29] свидетельствуют о том, что в некоторых случаях эти вирусы были образованы в ходе уникальных процессов рекомбинации.
В большинстве случаев несовпадения результатов с определением GT1 при полногеномном секвенировании было выявлено наличие GT6 (6/8), что указывает на то, что с помощью метода INNO-LiPA v2.0 все еще трудно дифференцировать эти два генотипа, хотя частота неверных результатов в этой версии ниже, чем в предыдущей [36, 38]. Сохранению проблемы в дифференцировке GT1 и GT6, возможно, способствует разнообразие и сложность последовательностей в подтипах GT6 даже при наличии зондов, направленных на ядро.
В целом уровень несоответствия результатов, требующих подтверждения, составлял 12,3 %, однако это касалось только подтипов GT2, 3, 4 и 6. Интересно, что наиболее высокая частота выявления новых/смешанных подтипов также отмечалась для GT2 (3,1 %), 4 (2,3 %) и 6 (4,7 %).
Очевидно, что разнообразие подтипов внутри каждого генотипа напрямую коррелировало с более высоким уровнем несоответствия результатов и необходимости в уточнении. Последовательности новых/смешанных подтипов не были полностью описаны, однако их обнаружение внутри данных генотипов указывает на то, что их разнообразие охвачено не до конца.
Глобальная распространенность подтипов подтвердила данные о широком разнообразии внутри подтипов GT2, 4 и 6. Высокое число циркулирующих подтипов GT2, 4 и 6 противоречит тому, что мы наблюдаем для GT1, 3 и 5, где преобладают подтипы GT1–2. Разнообразие и распространенность подтипов различались в зависимости от региона, что было показано в анализе распространенности генотипов [10].
Интересно, что во Франции были обнаружены новые/смешанные подтипы GT1 (n = 3; 0,8 %), 2 (n = 13; 28,3 %), 4 (n = 4; 3,4 %), 5 (n = 1; 1,8 %) и 6 (n = 1; 33,3 %). Причина повышения неописанных последовательностей во Франции неизвестна, хотя следует учитывать миграцию населения как фактор, способствующий их возникновению. Это явление подлежит изучению с помощью филогеографического анализа.
Подтипы вирусов и характер распределения МР являются репрезентативными в отношении пациентов, включенных в клинические исследования в конкретном медицинском центре конкретного региона, и могут не отражать общемировой картины. Возможно также, что на распределение подтипов повлияла миграция пациентов в центры проведения клинических исследований. Для оценки глобального распределения потребуется более обширный забор образцов для исследования.
Анализ МР показал, что резистентность к ПППД, направленных на NS5B, встречается нечасто, однако МР, направленные на NS3 и NS5A, регистрируются в популяции гораздо чаще. Из анализируемой популяции были исключены пациенты, уже получавшие ранее ПППД. При анализе последовательности NS5B МР L159F была обнаружена только у 8 % пациентов с вирусом GT1b (см. табл. 5).
Наличие мутации L159F связано со снижением частоты УВО только при использовании схем с небольшой продолжительностью (< 24 нед.), содержащих SOF и RBV, но не при применении комбинированных схем, направленных как на NS5A, так и на NS5B, содержащих PegIFN, или же при использовании рекомендуемого периода лечения [39]. МР S282T в последовательности NS5B, единственная МР с продемонстрированной in vitro резистентностью к SOF, не была обнаружена ни в одном из генотипов в начале исследования.
Варианты NS5A в положении МР Y93 были зарегистрированы во многих подтипах. Вариант Y93H был обнаружен в GT1b (11 %), GT3a (6 %), GT4b (50 %) и GT4r (13 %). GT4r характеризовался высокой распространенностью МР NS5A у 8 пациентов, включенных в анализ, у большинства из которых были обнаружены МР NS5A в начале исследования. В исследовании по применению ледипасвира (LDV)/SOF в течение 12 нед. у пациентов с инфекцией, вызванной HCV GT4, 2 из 3 пациентов с GT4r не достигли УВО [40]. У пациента с GT4r, у которого комбинация LDV/SOF оказалась эффективной, было 2 МР в NS5A (M28I + Q30R) в начале исследования, так же как и у большинства пациентов с GT4r в нашем анализе (50 %).
У 2 больных, у которых терапия оказалась неэффективной, имелось 3 МР в NS5A (M28M/V + Q30R + L31M) в начале исследования. В связи с небольшим объемом выборки остается неизвестным, обусловлена ли неудача в лечении более высоким числом МР в последовательности NS5A у пациентов с GT4r. Варианты последовательности NS5A, обнаруженные в вирусах GT1a и 1b, подтверждают результаты предыдущих публикаций [41].
МР в последовательности NS3 были обнаружены во множестве подтипов в анализируемой популяции. Варианты D168 были зарегистрированы во всех штаммах GT3a, 3b и 3i (D168Q), 53 % штаммов GT5a (D168E) и 7 % штаммов GT6a (D168E), а также у единичных пациентов со штаммами GT1c (D168E) и GT7 (D168Q). Вариант Q80K, с которым связана резистентность к симепревиру, обнаруживался с распространенностью 36 % у пациентов с HCV GT1a, а вариант Q80L был выявлен у 2 больных с GT1l.
Вариант V36L, который является причиной резистентности к телапревиру, наблюдался практически у всех пациентов с GT2–5. У пациентов с GT1c, 1e и 7 обнаруживались тройные комбинации МР в последовательности NS3. Применение комбинированных схем ПППД, а не монотерапии у таких больных может снизить влияние МР на исходы лечения.
В дальнейшем исследования должны быть направлены на изучение изменений распространенности подтипов HCV на фоне современного лечения, а также на оценку различий в ответе подтипов на существующие схемы ПППД, особенно с учетом специфических для подтипов полиморфизмов, выявленных в нашем анализе МР.
Различия в эффективности схем терапии на основании подтипов, очевидно, будут наиболее значимыми для GT2, 4 и 6, что окажет наиболее выраженное влияние на разработку схем лечения в Северной и Центральной Африке, на Ближнем Востоке и в Азии. Если подтипы внутри этих генотипов будут труднее поддаваться терапии, оптимизация диагностики и лечения будет ускорена за счет точного определения подтипов с помощью стандартных методов. Ключевое значение имеет непрерывное наблюдение с целью разработать наиболее эффективные стратегии элиминации HCV во всем мире.
Спонсоры
Финансовая поддержка была оказана компанией Gilead Sciences, Inc.
Конфликты интересов
Д-ра Bhardwaj, Hedskog, Camus, McNally, Brainard, Miller, Mo и Svarovskaia являются сотрудниками и держателями акций компании Gilead Sciences, Inc. Д-р Chodavarapu — держатель акций компании Gilead Sciences, Inc. Д-р Welzel — консультант и спикер в компаниях AbbVie, Boehringer-Ingelheim, Bristol-Myers Squibb, Janssen и Gilead Sciences, Inc. Д-р Zeuzem — консультант в компаниях AbbVie, Bristol-Myers Squibb, Janssen, Merck/MSD и Gilead Sciences, Inc. Д-р Jacobson — спикер и/или консультант в компаниях AbbVie, Achillion, Bistrol-Myers Squibb, Enanta, Janssen, Merck, Tobira и Gilead Sciences, Inc. Д-р Agarwal — спикер и/или консультант в компаниях AbbVie, Achillion, Astellas, Bristol-Myers Squibb, GlaxoSmithKline, Janssen, Merck, Novartis и Gilead Sciences, Inc.
Все подробности смотрите в дополнительном файле раскрытия конфликтов интересов ICMJE.
Вклад авторов
Концепция и дизайн: TW, NB, CH, KC, GC, JM, DB, MM, HM, ES, IJ, SZ, KA.
Эксперименты и процедуры: TW, NB, CH, KC, GC, JM, DB, MM, HM, ES, IJ, SZ, KA.
Написание рукописи и обзор: TW, NB, CH, KC, GC, JM, DB, MM, HM, ES, IJ, SZ, KA.
Благодарности
Мы выражаем благодарность пациентам, принимавшим участие в клинических исследованиях II–III фазы, а также персоналу компании Gilead.
Дополнительные материалы
Дополнительные материалы к статье представлены в англоязычном приложении на сайте https://dx.doi.org/10.1016/j.jhep.2017.03.014.
Литература
- Shepard CW, Finelli L, Alter MJ. Global epidemiology of hepatitis C virus infection. Lancet Infect Dis 2005;5:558–567.
- Lauer GM, Walker BD. Hepatitis C virus infection. N Engl J Med 2001;345:41–52.
- Smith DB, Bukh J, Kuiken C, Muerhoff AS, Rice CM, Stapleton JT, et al. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: updated criteria and genotype assignment web resource. Hepatology 2014;59:318–327.
- Timm J, Roggendorf M. Sequence diversity of hepatitis C virus: implications for immune control and therapy. World J Gastroenterol 2007;13:4808–4817.
- Asselah T, Boyer N, Saadoun D, Martinot-Peignoux M, Marcellin P. Direct-acting antivirals for the treatment of hepatitis C virus infection: optimizing current IFN-free treatment and future perspectives. Liver Int 2016;36:47–57.
- Sulkowski MS, Gardiner DF, Rodriguez-Torres M, Reddy KR, Hassanein T, Jacobson I, et al. Daclatasvir plus sofosbuvir for previously treated or untreated chronic HCV infection. N Engl J Med 2014;370:211–221.
- Lawitz E, Mangia A, Wyles D, Rodriguez-Torres M, Hassanein T, Gordon SC, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med 2013;368:1878–1887.
- Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med 2015;373:2599–2607.
- Foster GR, Afdhal N, Roberts SK, Brau N, Gane EJ, Pianko S, et al. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med 2015;373:2608–2617.
- Gower E, Estes C, Blach S, Razavi-Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol 2014;61:S45–S57.
- Messina JP, Humphreys I, Flaxman A, Brown A, Cooke GS, Pybus OG, et al. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015;61:77–87.
- Rehman IU, Idrees M, Ali M, Ali L, Butt S, Hussain A, et al. Hepatitis C virus genotype 3a with phylogenetically distinct origin is circulating in Pakistan. Genet Vaccines Ther 2011;9:2.
- McNaughton AL, Cameron ID, Wignall-Fleming EB, Biek R, McLauchlan J, Gunson RN, et al. Spatiotemporal reconstruction of the introduction of hepatitis C virus into Scotland and its subsequent regional transmission. J Virol 2015;89:11223–11232.
- Akkarathamrongsin S, Hacharoen P, Tangkijvanich P, Theamboonlers A, Tanaka Y, Mizokami M, et al. Molecular epidemiology and genetic history of hepatitis C virus subtype 3a infection in Thailand. Intervirology 2013;56:284–294.
- Karchava M, Waldenstrom J, Parker M, Hallack R, Sharvadze L, Gatserelia L, et al. High incidence of the hepatitis C virus recombinant 2k/1b in Georgia: Recommendations for testing and treatment. Hepatol Res 2015;45:1292–1298.
- Raghwani J, Thomas XV, Koekkoek SM, Schinkel J, Molenkamp R, van de Laar TJ, et al. Origin and evolution of the unique hepatitis C virus circulating recombinant form 2k/1b. J Virol 2012;86:2212–2220.
- Kalinina O, Norder H, Mukomolov S, Magnius LO. A natural intergenotypic recombinant of hepatitis C virus identified in St Petersburg. J Virol 2002;76:4034–4043.
- Wyles DL. Antiviral resistance and the future landscape of hepatitis C virus infection therapy. J Infect Dis 2013;207:S33–S39.
- Paolucci S, Fiorina L, Mariani B, Gulminetti R, Novati S, Barbarini G, et al. Naturally occurring resistance mutations to inhibitors of HCV NS5A region and NS5B polymerase in DAA treatment-naive patients. Virol J 2013;10:355.
- Sarrazin C, Lathouwers E, Peeters M, Daems B, Buelens A, Witek J, et al. Prevalence of the hepatitis C virus NS3 polymorphism Q80K in genotype 1 patients in the European region. Antiviral Res 2015;116:10–16.
- Stuyver L, Rossau R, Wyseur A, Duhamel M, Vanderborght B, Van Heuverswyn H, et al. Typing of hepatitis C virus isolates and characterization of new subtypes using a line probe assay. J Gen Virol 1993;74:1093–1102.
- Stuyver L, Wyseur A, van Arnhem W, Lunel F, Laurent-Puig P, Pawlotsky JM, et al. Hepatitis C virus genotyping by means of 5′-UR/core line probe assays and molecular analysis of untypeable samples. Virus Res 1995;38:137–157.
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25:3389–3402.
- Hedskog C, Chodavarapu K, Ku KS, Xu S, Martin R, Miller MD, et al. Genotype- and subtype-independent full-genome sequencing assay for hepatitis C virus. J Clin Microbiol 2015;53:2049–2059.
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994;22:4673–4680.
- Zwickl DJ. Genetic algorithm approaches for the phylogenetic analysis of large biological sequence datasets under the maximum likelihood criterion Ph.D. dissertation. The University of Texas at Austin; 2006.
- Anisimova M, Gascuel O. Approximate likelihood-ratio test for branches: A fast, accurate, and powerful alternative. Syst Biol 2006;55:539–552.
- Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 2010;59:307–321.
- Hedskog C, Doehle B, Chodavarapu K, Gontcharova V, Crespo Garcia J, De Knegt R, et al. Characterization of hepatitis C virus intergenotypic recombinant strains and associated virological response to sofosbuvir/ribavirin. Hepatology 2015;61:471–480.
- Schreiber J, McNally J, Chodavarapu K, Svarovskaia E, Moreno C. Treatment of a patient with genotype 7 HCV infection with sofosbuvir and velpatasvir. Hepatology 2016;64(3):983–985.
- Frank C, Mohamed MK, Strickland GT, Lavanchy D, Arthur RR, Magder LS, et al. The role of parenteral antischistosomal therapy in the spread of hepatitis C virus in Egypt. Lancet 2000;355:887–891.
- Lin MV, Chung R. Recent FDA approval of sofosbuvir and simeprevir. Implications for current HCV treatment. Clin Liver Dis 2014;3:65–68.
- Kumada H, Suzuki Y, Ikeda K, Toyota J, Karino Y, Chayama K, et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology 2014;59:2083–2091.
- Nakamoto S, Kanda T, Wu S, Shirasawa H, Yokosuka O. Hepatitis C virus NS5A inhibitors and drug resistance mutations. World J Gastroenterol 2014;20:2902–2912.
- Lam AM, Espiritu C, Bansal S, Micolochick Steuer HM, Niu C, Zennou V, et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob Agents Chemother 2012;56:3359–3368.
- Bouchardeau F, Cantaloube JF, Chevaliez S, Portal C, Razer A, Lefrere JJ, et al. Improvement of hepatitis C virus (HCV) genotype determination with the new version of the INNO-LiPA HCV assay. J Clin Microbiol 2007;45:1140–1145.
- De Keukeleire S, Descheemaeker P, Reynders M. Diagnosis of hepatitis C virus genotype 2k/1b needs NS5B sequencing. Int J Infect Dis 2015;41:1–2.
- Noppornpanth S, Sablon E, De Nys K, Truong XL, Brouwer J, Van Brussel M, et al. Genotyping hepatitis C viruses from Southeast Asia by a novel line probe assay that simultaneously detects core and 5’ untranslated regions. J Clin Microbiol 2006;44:3969–3974.
- Svarovskaia ES, Gane E, Dvory-Sobol H, Martin R, Doehle B, Hedskog C, et al. L159F and V321A sofosbuvir-associated hepatitis C virus NS5B substitutions. J Infect Dis 2016;213:1240–1247.
- Abergel A, Metivier S, Samuel D, Jiang D, Kersey K, Pang PS, et al. Ledipasvir plus sofosbuvir for 12 weeks in patients with hepatitis C genotype 4 infection. Hepatology 2016;64(4):1049–1056.
- Zeuzem S, Mizokami M, Pianko S, Mangia A, Han KH, Martin R, et al. NS5A resistance-associated substitutions in patients with genotype 1 hepatitis C virus: prevalence and effect on treatment outcome. J Hepatol 2017;66:910–918.
Имеются противопоказания! Необходима консультация специалиста
